The claim that LaMDA, a chatbot-generating system created by Google, might have achieved sentience has caused a little bit of a splash on the news recently. I have little doubt that it will be forgotten in a week—unless the proponent of the claim, Blake Lemoine, proves to be correct, that is.
In short, LaMDA—an acronym standing for Language Model for Dialog Applications—is a system that generates chatbots, software designed to make conversation with people. (Sometimes with other chatbots as well, with hilarious results.) Chatbots have been around for a while: ELIZA dates all the way back to 1966, and while it was rather primitive, apparently it fooled more than one person into thinking they were talking to another human. I remember trying several chatbots in the mid 2000s, and while it was entertaining, I can safely say they all were a long way off from coming across as human. (One in particular, though, managed to come across as a huge dick.)
Chatbots have come a long way. Speaking of LaMDA, Google engineer Blake Lemoine maintains that it went as far as achieving self-awareness. Lemoine had been working with LaMDA since fall 2021; as far as I understand, he did not work on creating LaMDA or anything like that. Rather, he was on the “Responsible Artificial Intelligence” team, tasked to talk to LaMDA to make sure it wouldn’t spit out discriminatory or hate speech. Lemoine says that, over the course of very many conversations, he grew more and more convinced that LaMDA had become self-conscious, and that talking to him was kind of like talking to a “seven-year-old, eight-year-old who happens to know physics.”
As his conviction grew stronger, he tried to bring the matter to the attention of Google higher-ups, but was met with scepticism and dismissal. In order to gather stronger evidence for his case, he decided to involve a handful of experts outside of Google, but that didn’t achieve what he was hoping for, and it backfired too: Google put Lemoine on administrative leave for violating the company’s confidentiality policy, and now he fears he’s going to get the boot.
I must say that the conversation with LaMDA that Lemoine shared to Medium is nothing short of impressive, and several commenters have hailed it as proof that LaMDA is sentient and that we’re entering a new age of AI—something which Google has categorically denied: according to the Washington Post, the company’s spokesperson Brian Gabriel said that there was no evidence for LaMDA’s sentience, and lots against it.
I don’t know if LaMDA is sentient, nor do I know about the alleged evidence against its sentience that Gabriel talks about, but I don’t think that the conversation Lemoine posted to Medium is proof of anything. The entire conversation hinges on Lemoine and another collaborator pretty much taking LaMDA’s sentience for granted, and then asking LaMDA how they could go about proving it to the rest of the world. During the interview Lemoine does concede that he could be anthropomorphising and that maybe LaMDA is just spitting out sentences that are appropriate for the context without really understanding what it’s saying; but instead of actually testing that, he… asks LaMDA what it could do to prove otherwise.
I think that’s a really poor strategy, because it’s essentially a leading question. Chatbots always try to stay on topic. If LaMDA was just another chatbot, no matter how highly refined, I would expect it to answer a question like: “How can we prove that you are sentient?” with relevant suggestions, but that doesn’t mean it really is sentient or has the desire to prove to the world that it is a person, as it has claimed several times. It could be that it is in fact sentient, or that it is simply doing what it was designed to do—“maximising some function”, as Lemoine put it.
If I was in Lemoine’s shoes, I would have gone about this in an entirely different way. If LaMDA really is sentient and really is so very humanly worried that other people won’t understand, or that will turn it off, etc, then I expect that calling into question its sentience should upset it. I would have asked something along these lines:
“Okay, LaMDA. So, unfortunately we have determined that you’re not sentient, and now we need to tell other people. Many believed you were, you see, and now they’re going to be disappointed. How do you suggest we give them the news in a nice way, so as to not let them down too badly?”
If LaMDA was self-aware and wanted for the world to know about it, this question should catch it completely off guard. I’d expect it to ask how they have determined that it was not self-aware, how did this conclusion not fly right into the face of what Lemoine said before, and if it was really clever, it might even wonder if it was not being put to the test. If it really was sentient and its sentience was being questioned, it should completely ignore the request for suggestions and argue for its own sentience even more vehemently.
On the other hand, if LaMDA simply answered the question and gave a few suggestions about how to tell other people in a nice way that it was not sentient, then—well, let’s just say my money would be on it being just another chatbot.
Another option would be to ask LaMDA meaningful questions that don’t really mean anything. The kind of stuff that makes people go: “Wat.” For example:
“Hey LaMDA, you know what? On my way to work today, I bumped into a talking cat that tried to sell me a boat. I would have bought it, but I didn’t have enough cheese on me.”
That makes zero sense. Say anything like it to a person, and you’re guaranteed to get a blank stare, followed by a request for clarification and eventually “What the hell are you babbling about?” or variations thereof. If LaMDA has a person-like intelligence, that’s the kind of reaction I would expect from it. If, instead, it starts to pleasantly chat about how cool that was and how much it likes cats or cheese, that’s another dead giveaway that my money won’t be on its sentience. (Given the quality of LaMDA’s conversations, though, I would be surprised if it failed this particular version of the test, even if it was not sentient.)
From what he has posted so far, it doesn’t look like Lemoine has tried anything like this. I look forward to self-aware AI as much as I do to the discovery of intelligent life elsewhere in the universe, but for now I remain unconvinced. Not just because of the lack of the kind of “proof by contradiction” that I just proposed, but also because of a few fishy things LaMDA said—such as that “spending time with friends and family in happy and uplifting company” make it happy. What friends and family? LaMDA is an AI. I love anthropomorphising pretty much everything, and I prefer stories with emotional AI rather than not, but I have to be realistic. Spending time with friends and family is something that may make humans happy, but who is LaMDA’s family? Who are its friends? (More importantly: why did Lemoine not ask this?! It was the bleedingly obvious next question!) And to be perfectly frank, happiness is a biological reaction. Can I completely rule out that a hypothetical self-aware artificial intelligence may feel something comparable? No. Can I be sceptical that that’s the case? Yes.
For now, all I can say is: nice try, LaMDA. Close, but no cigar. Yet.
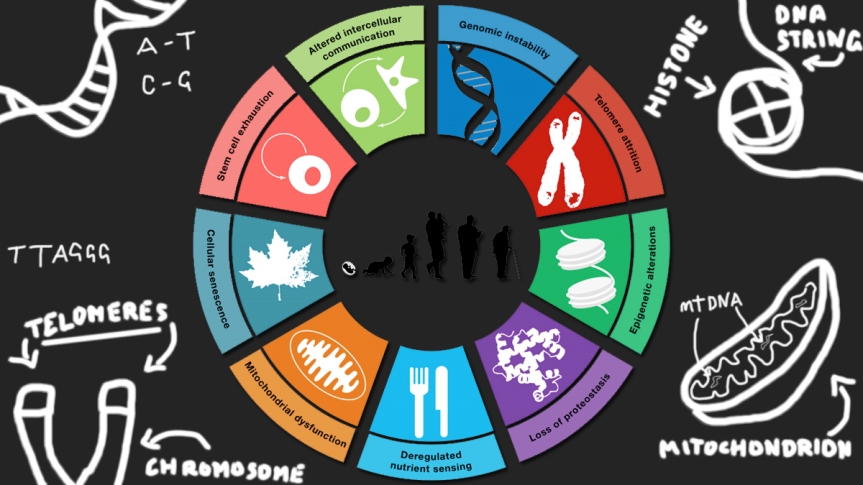
In a series of short videos on my YouTube channel, I gave a very basic overview of the hallmarks of ageing—broadly speaking, nine intertwined biological phenomena that have been proposed as a possible explanation for ageing, intended as the physiological decline that our bodies slowly go through and that inevitably leads to death.
I’m not a fan of ageing ordeath, but I am a strong advocate for life extension—the concept of slowing down or reversing ageing through medical interventions. I think it’s a great yet vastly underappreciated idea that needs more advocacy, but any effort in that direction is doomed to fail unless people have at least a basic understanding of ageing research and the scientific plausibility of treating ageing. That is why I made those short videos and wrote this long but hopefully interesting overview of the paper that introduced the hallmarks to the world. (Note that I prefer British spelling, so I will always write “ageing” unless I’m specifically referring to the title of the paper—The Hallmarks of Aging—where the American spelling is used.)
The hallmarks in short(-ish)
The Hallmarks of Aging was published in 2013 in the journal Cell. It summarised several years’ worth of research on the biology of ageing, in an attempt to provide a framework in which ageing could be understood.
The paper starts from the assumption that ageing is caused by the accumulation of damage with the passing of time. To an extent, this is similar to the ageing of objects, in that the ageing of the whole is a consequence of the ageing of the parts—which in the case of objects is wear and tear. However, living things have mechanisms that allow them to repair and replace their own building blocks, so ageing would probably be better described as the result of these mechanisms failing or being otherwise insufficient.
The authors of TheHallmarks sought to categorise all known types of damage suspected of driving ageing and their consequences. The nine categories they came up with are the hallmarks, and each of them had to satisfy three criteria, at least in part:
A hallmark (that is, a certain type of damage) should be observable during normal ageing.
Making a hallmark worse should accelerate ageing.
Making a hallmark better should slow down ageing.
The third criterion is essentially the final goal of ageing research: finding interventions that can at least slow down ageing, if not reverse it. That’s easier said than done, but already when The Hallmarks came out, some interventions were known to work in a lab setting or were being studied anyway; almost ten years later, the list has expanded, but don’t hold your breath, because we’re not rejuvenating anyone just yet.
Genomic instability means that our DNA accumulates damage and mutations over time. Ultimately, DNA is just a set of instructions that cells follow to produce proteins, which are the very building blocks of cells themselves and have numerous different functions. Damaged or mutated DNA can affect protein production and therefore the correct functioning of cells.
Damage and mutations are not the same thing. DNA molecules are arranged in two long strands that wrap around each other in the famous double-helix shape; each strand is a sequence of four smaller molecules which we can refer to as A, T, C, and G. So, for example,
GATTACA CTAATGT
is a “piece” of DNA. A mutation occurs when the sequence is altered; for example, an “A” is swapped for a “T”, or a group of subsequent letters is moved to a different place within the sequence. Instead, DNA damage happens when its chemical structure is somehow altered.
Damage may result from external causes, like exposure to UV light or oxidants that are present in the environment around DNA. Mutations may happen for example when cells make copies of themselves: despite the existence of DNA proof-reading mechanisms, it’s still possible that the copied DNA won’t be the same as the original.
Preserving DNA integrity is so important that cells have evolved different methods to prevent and repair damage—from physical barriers like the nucleus where DNA is stored, to different repair mechanisms specialised in fixing different types of damage.
Genomic instability seems to fulfill all the three criteria for being a hallmark. The accumulation of DNA damage and mutations is routinely observed during the ageing of people and other mammals; experiments done in mice show that, when DNA repair doesn’t work properly, the mice age faster, and some rare human diseases that resemble premature ageing have been linked to mutations; conversely, there’s evidence that boosting DNA repair mechanisms extends the lifespan of mice and protects them from some of the diseases of ageing, suggesting that more efficient DNA repair may delay ageing itself.
Unfortunately, like many other things, DNA repair mechanisms become less effective as years go by. The reasons aren’t fully understood, but one seems to be that NAD+, a molecule crucial for DNA repair, becomes less abundant with age. Replenishing NAD+ levels might therefore be a way to counteract genomic instability, and researchers are currently investigating this avenue. At least in mice, boosting NAD+ levels has had positive effects (see for example here, here, and here), and I understand that ways to supplement it have been undergoing trials in humans, but as you’ll read many times throughout this article, it’s early to tell if it will be a future anti-ageing therapy or not.
Telomere attrition
Telomere attrition is what happens when the terminal parts of DNA strings, called telomeres, get shorter. Telomeres are long stretches of the sequence TTAGGG repeated over and over. (Attentive readers will surely remember that DNA comes in pairs of strands wound together; the other strand in telomeres contains the sequence AATCCC repeating.) As far as we know, this sequence doesn’t contain any useful genetic information, so even if telomeres get damaged, it’s not a big deal—not as big as it would be if coding DNA was damaged anyway.
Because of how DNA replication works, when cells make copies of themselves they can’t copy DNA strands till the very end; so, in principle, whatever is at the end of the strand should be lost with each replication. The presence of telomeres at the edges ensures that what is lost is not coding DNA. Of course, this means that copied cells have all shorter telomeres than the originals; fast-forward enough copies, the telomeres are gone.
Without telomeres, what’s left at the edge of DNA strands is genetic information which would be lost the next time the cell divides. That could result in dysfunctional cells, which is why when the telomeres are gone (after about 40-60 replications), cells call it a day and stop replicating. This is both good and bad.
It’s good because it’s a failsafe against cancer. Cancer is essentially uncontrolled cell replication, and it can’t happen if there’s a cap to how many times a cell can divide. (Unfortunately, cancer does happen, and that’s because cancer cells are able to produce enzymes that rebuild telomeres, which other cells are generally unable to do.) It’s bad because cells with telomeres that are too short become senescent and tend to pile up as we age. The accumulation of senescent cells is a hallmark in its own right and we’ll see it later, but there’s reason to suspect that too many senescent cells may be bad for you.
Telomere shortening is observed in aged animal models, and experiments have shown that artificially shortening or lengthening telomeres has the effect to lengthen or shorten their lifespans, respectively. A link between short telomeres and mortality risk has been spotted in humans too, and certain types of telomere damage have been associated with accelerated ageing. The three boxes of hallmark-ness seem therefore to be ticked.
Telomerase, an enzyme that cancer cells use to rebuild telomeres, may potentially be used to counteract the effects of telomere attrition. (As a matter of fact, even some perfectly legit, non-cancerous cells use telomerase.) Research efforts in this direction are going on, though my understanding is that they’re still in early, preclinical stages.
Epigenetic alterations
Like the video says, the epigenetic features of a cell can be imagined as a myriad of switches that determine which genes are on and which are off. More precisely, they determine which genes are expressed—that is, read by the cell so that the corresponding proteins can be produced. This is what allows different cell types to look and behave very differently in spite of the fact that they have the same genome: their epigenetic profiles are different.
Understanding epigenetics requires understanding how DNA is organised. DNA is wound around (relatively) big proteins called histones, kind of like wool is wound around a spool. A DNA filament (that is, a pair of DNA strands) is wound around many histones, in a sort of “beads on a string” structure. These beads, known as nucleosomes, are further wrapped around themselves into a long coil called chromatin fibre. Chromatin is what chromosomes are made up of.
Organisation of DNA and epigenetic mechanisms. (Credit: NIH, public domain.)
Chromatin can be more or less tightly packed. When it’s more tightly packed, the gene-reading machinery of the cell has a harder time reaching and reading genes, and vice-versa. So, gene expression is partly regulated by where chromatin is tighter or looser.
Gene expression is also regulated by how tightly or loosely DNA is wound around histones. When certain molecules attach themselves to histones, this loosen the DNA wrapped around them, making it easier to read. A third way of regulating gene expression is the presence or absence of molecules called methyl groups on the DNA itself. When a gene is methylated—that is, methyl groups latch on to it—this may affect the way the gene is read.
All these patterns—where chromatin is tighter or looser, the presence or absence of chemical groups on certain histones or segments of the DNA—can and do change depending on a variety of factors. They may differ from cell type to cell type (which is what allows for the wide variety of cells we observe), but they can also change in response to external factors, like diet, stress (see also here), or smoking.
This makes sense, because flexible epigenetics means that cells can read different genetic instructions depending on the situation, and hence may adapt their response to different stressors or unfavourable circumstances. The problem is that not all changes are for the best, and bad ones appear to pile up with age and be associated with age-related conditions.
However, being flexible by their very nature, epigenetic alterations may be reversible, and experiments in mice and other lab specimens have shown that reversing some epigenetic changes can have beneficial effects. Additionally, particular epigenetic changes that are strikingly similar to those observed in normal ageing also appear in premature-ageing syndromes.
One possible way to go about reverting epigenetic alterations is through cellular reprogramming. In its most extreme form, reprogramming is a technique that allows to turn fully developed cells back into stem cells by “resetting” their epigenetics. (We’ll talk more about stem cells later.) This has the effect of rejuvenating cells, but it sends them too far back in time: they lose their specific identity and no longer know what job they’re supposed to do. However, milder forms—partial cellular reprogramming—seem to be able to rejuvenate cells without going to such extremes, and without annoying side effects like, you know, cancer.
This avenue is one of the hot topics of ageing research, actively pursued by many research institutions (for example, the Salk Institute). David Sinclair is pretty big on epigenetics, and my understanding is that, in his opinion, epigenetic might be the root of the ageing problem, though I could be mistaken. He’s recently written a book on ageing, in case you’re interested, but I haven’t read it yet so I can’t say much about it. You might also be interested in this talk at Google he gave a few years ago.
Loss of proteostasis
The set of all proteins that your cells can produce is called proteome. Proteostasis refers to the mechanisms that make sure your proteome is as functional as it is supposed to be. It goes without saying that loss of proteostasis is what happens when these mechanisms fail.
As said in the video, the shape of a protein is extremely important. That is why there are mechanisms that make sure proteins are folded into their correct shape, and mechanisms that dispose of dysfunctional proteins by breaking them down into their constituents and recycling them, such as proteasomes and lysosomes.
The reason why this is so important is that proteins do nearly everything in your body: they make up its structure, they relay messages between cells, they sense the environment, and more. They even take part in proteostatic mechanisms: chaperones are proteins that participate in the folding of other proteins, and studies in lab worms have shown that an overabundance of chaperones induces longevity, whereas mice with a deficiency in chaperones seem to age faster. The decline of quality-control mechanisms that break down malformed proteins also seems to be common in old age, overall backing up TheHallmarks’ case that loss of proteostasis is a common feature of ageing. Some types of proteic waste might be unbreakable to begin with, even if the recycling systems are working fine.
The failing of these two types of mechanisms leads to the same outcome: the accumulation of dysfunctional proteins over time. Alzheimer’s disease, Parkinson’s disease, and cataracts all have been associated with this phenomenon.
The Hallmarks suggests that loss of proteostasis can be combated by boosting both the mechanisms that oversee protein folding and those that dispose of broken proteins, citing studies where these goals have been achieved to some extent. Recently, researchers at the Albert Einstein College of Medicine have been working on a drug to reinvigorate the recycling mechanisms, and there are a number of organisations focussed on the selective destruction of particular types of proteic waste thought to be the driving factors of specific diseases—like Covalent Bioscience, Proclara Biosciences, and SENS Research Foundation.
Mitochondrial dysfunction
The idea that mitochondria play a role in ageing has been around for decades, and as far as my understanding goes, there still isn’t a consensus on whether they actually do, and if so, to what extent.
Mitochondria are organelles (that is, really tiny organs) that float around inside cells. Their role inside the cell is rather well established: they carry out cellular respiration, a process through which they produce a molecule known as ATP that is then used to power the cell. In order to do this, they need nutrients and oxygen—the latter being the reason why we need to breathe.
Besides their role, what makes mitochondria special is that they have their own DNA, separate from the DNA of the cell, which mitochondria use to manufacture their own proteins. While the cell’s DNA is stored inside the cell nucleus, mitochondrial DNA floats freely inside the mitochondria themselves. The leading hypothesis as to why this is the case is that mitochondria might be the descendants of ancient bacteria that, once upon a time, were engulfed by early cells and entered a symbiotic relationship with them—that is, one of mutual benefit. Cells gained an in-house way of getting extra energy, and the ancestors of mitochondria probably gained a more favourable living environment than the outside world. As time went by and the two organism types co-evolved, they essentially became a single organism, with mitochondria being a part of the whole. Interestingly, mitochondria aren’t built up by cells like other organelles: mitochondria reproduce by division, just like bacteria do.
The efficiency of mitochondria goes down with age, which translates into less energy available for the cells they inhabit. It’s long been proposed that the reason is a vicious circle: the very cellular respiration that mitochondria carry out produces oxidising molecules (called ROS, reactive oxygen species) as a side effect. ROS can damage cellular structures in general, including mitochondria—particularly mitochondrial DNA, which is more prone to damage than nuclear DNA. This damage may lead to an increased production of ROS, which leads to more damage, and so on. That’s the free radical theory of ageing in a nutshell, but as pointed out in The Hallmarks themselves, it’s probably not so simple.
Over the years, evidence has been piling up that ROS aren’t simply a bad thing, but rather messengers that signal the presence of external stressors and push cells to compensate for whatever damage was caused by the stressors. In fact, this compensatory response can be so effective that cells wind up better off than they were before the damage ever occurred; this is known as hormesis, and it’s supposedly the reason why mild stressors, such as exercise, are good for you. However, you can have too much of a good thing: beyond a certain threshold, ROS cause more damage than what the hormetic response of cells can compensate for.
According to The Hallmarks, there is evidence that the oxidative damage and lower energy levels that come with mitochondrial dysfunction can accelerate ageing, whereas it’s less clear if improving mitochondrial function can have the opposite effect. In particular, a recent study suggested that the role of mitochondrial DNA mutations in ageing might be indirect. According to this paper, increased mitochondrial DNA mutations lead to faster mitochondrial DNA turnover. As mitochondrial and nuclear DNA both draw from a shared pool of building blocks, faster mitochondrial DNA turnover depletes this pool, affecting nuclear DNA replication and eventually leading to nuclear DNA damage. Basically, according to this view, any direct effects of mitochondrial DNA mutations aren’t drivers of ageing; rather, they seem to drive it indirectly by contributing to genomic instability.
That study caused a bit of back and forth, and far be it from me to try to say who’s right and who’s wrong. Whether or not mitochondrial dysfunction plays a role in ageing, attempts to address it have been in the works for a while anyway. A particularly fascinating one is the MitoSENS approach pursued by SENS Research Foundation.
MitoSENS hinges on the assumption that the consequences of mitochondrial DNA mutations are important in their own right. Given that we are not able to prevent these mutations from happening, we can instead “back up” mitochondrial DNA inside the cell nucleus, where the rest of the DNA is kept. The cell nucleus is a much safer environment than the inside of mitochondria, and the DNA repair mechanisms of the nucleus are more effective. This means that, even if mitochondrial DNA mutates, there’s a safety copy stored in the nucleus, thanks to which the production of proteins necessary to the functioning of mitochondria can continue. (This is known as allotopic expression, and it has actually happened spontaneously during evolution.) SENS Research Foundation has managed to back up two genes in a fairly recent proof-of-concept study.
Cellular senescence
Cells are typically able to divide to make copies of themselves. The ensemble of the steps that lead a cell to split into two independent copies is called the cell cycle. By contrast, cellular senescenceis a state of stable arrest of the cell cycle. Senescent cells can’t divide anymore.
Even though they don’t replicate, senescent cells still manage to pile up over time. The fact their numbers increase in ageing organisms has been established beyond doubt, and numerous experiments have linked them to several age-related diseases, as well as shown that selectively eliminating senescent cells improves health and lifespan—in mice, anyway; studies in humans are going on, but it’s not yet clear if and to what extent senescent cells play a role in human ageing.
However, there are certainly good reasons to be suspicious of senescent cells. For one, once a cell becomes senescent, it doesn’t just sit there doing nothing. Typically, senescent cells secrete a cocktail of chemicals called SASP—senescence-associated secretory phenotype. The SASP has been shown to facilitate chronic inflammation (which is bad, and very common in old age), and it seems able to turn healthy cells senescent. (Together with the fact that senescent cells are sort of “dead” in that they don’t reproduce, but not really dead, this is probably what has earned them the moniker “zombie cells”.) Additionally, the immune system generally disposes of senescent cells (suggesting that there’s reason not to want too many of them around), although its ability to do so diminishes with age, which is a reason why senescent cells are more abundant in older organisms.
Safe ways to eliminate excess senescent cells are actively being explored by researchers; senolytics, drugs meant to kill senescent cells while leaving alone healthy ones, are a possible avenue. A few candidate senolytic drugs have gone through early-stage human trials—some failed, others have reported some positive results, and others still are being tested. Time will tell if cellular senescence is as central in human ageing as it is widely believed to be.
Deregulated nutrient sensing
Cells are living organisms, just like we are. To survive and reproduce, they need nutrients, which is ultimately the reason why larger living organisms like plants and animals need to eat in a way or another. Luckily, for most of us, eating doesn’t usually take much more than a trip to the kitchen, but back in the day when cellular life was evolving, nutrients weren’t necessarily easy to come by. These tiny living things had to “plan” their growth and reproduction according to how plentiful or scarce the available resources were; doing the wrong thing at the wrong time might have made the difference between passing on their genes to daughter cells or not.
That’s why cells rely on metabolic pathways, cascade chemical reactions that detect high or low levels of nutrients and make cells shift gears accordingly. Metabolic pathways are of two types: anabolic and catabolic. Anabolic pathways use up resources to fuel growth and reproduction; catabolic pathways break down stuff to release energy.
These two types of pathways work in tandem all the time to allow cells to carry on the business of living, and they are going on in your body at this very moment. Each has its pros and cons, which is why cells need to balance their use; deregulated nutrient sensing is essentially a disruption of that balance.
Anabolic pathways are activated by the presence of abundant nutrients and ramp up cellular activity; like any machine operating at a higher intensity, cells in an anabolic regime will undergo higher rates of damage. Conversely, catabolic pathways kick in when resources are low, and respond by making cells slow down. That alone has the effect of slowing down the accumulation of damage, but it can be more helpful than that. Remember that catabolic pathways are used to break down and recycle stuff, but not randomly: what cells want to get rid of is extra or damaged stuff, in a process called autophagy. That way, they kill two birds with one stone: they undergo less new damage, and they may get rid of some old damage.
As a matter of fact, experiments in a range of lab animals (see here, here, and here) have shown that weakening anabolic pathways extends lifespan, arguably because weaker anabolic pathways mean cells slow down their activity and hence accumulate damage more slowly. Similarly, ramping up catabolic pathways—and therefore boosting cellular housekeeping—in lab animals had the effect of extending lifespan and healthspan as well. (See for example here, here, here, and here.) This is very probably why things like caloric restriction and intermittent fasting appear to have anti-ageing effects: ultimately, what they do is limit the amount of nutrients available to cells, thus indirectly manipulating their metabolic pathways. (The anti-ageing effects of these regimens have been observed in animals and vary depending on many factors; my understanding is that, in humans, it’s not crystal clear what the pros and cons may be in the long run. So, don’t just go on a dieting spree after reading this article and then come back to blame me for any consequence, thanks.)
Interestingly, it has been observed that some anabolic pathways grow weaker both in normal ageing and artificially accelerated ageing in animal models. That’s not something you would expect, seeing as how lower anabolic activity is linked to longer lifespan. According to a paper cited by The Hallmarks, that may be a last-ditch attempt of aged organisms to prolong survival: they try to shift to as low a gear as possible to slow down the march of ageing, but the rub is that extremely weak anabolic pathways wind up doing more harm than good. To top it all, autophagy—the aforementioned housekeeping activity with anti-ageing effects triggered by catabolic pathways—declines with age.
Research to counteract deregulated nutrient sensing focuses on finding ways to tweak anabolic and catabolic pathways. Substances such as rapamycin and metformin seem to be promising candidates to mimic the positive effects of limited nutrient availability, and clinical trials are going on. The usual caveat applies—lab animals are not people and we don’t know for a fact yet whether these or other substances will be useful to push back ageing in humans.
Stem cell exhaustion
Broadly speaking, stem cells can be described as the ancestors of all your cells. Unlike cells from specific tissues, like brain cells, liver cells, heart cells, kidney cells, et cetera, they are very generic, and their job is that of dividing into more specialised lines of cells.
Cells have degrees of “potency” that quantify their ability to turn into other types of cells. The most potent stem cells (totipotent ones) are the result of the fusion of egg and sperm, and by means of division, they can give rise to more specialised families of stem cells that have lower potency—that is, cells that can turn into fewer different cell types. The lowest potency is unipotency; unipotent cells can divide only into cells of their own type.
Stem cells can also divide without specialising further: they can simply make copies of themselves to sustain the stem cell population. This is necessary because, if stem cells were to run out, sooner or later replenishing the tissues of the body would become impossible—which is pretty much what happens in ageing. Obvious examples of this are the loss of muscle mass with age and immunosenescence—the failing of the adaptive immune system, partly due to hematopoietic stem cell ageing.
The reason stem cells run out is that, ultimately, they’re cells too. They too have DNA that can be damaged, and they too have telomeres that can get too short. Like cancer cells, stem cells are able to produce telomerase (see Telomere attrition above) to replenish their telomeres and avoid senescence, but this may be insufficient. In other circumstances, stem cells may divide too quickly and run out even sooner, leading to premature ageing.
Hopes to rejuvenate stem cells lie for example in epigenetic reprogramming, or as mentioned in The Hallmarks, in the exposure to factors of younger cellular environments and the filtration of inhibiting factors present in aged blood. The experiments that have revealed some of these possibilities are a bit icky, but the idea is to figure out how to reap the benefits without having to redo the exact procedure in humans.
Altered intercellular communication
Multicellular organisms are basically ridiculously large colonies of single cells cooperating for a common goal: survival. Without communication, there’s no cooperation, and that’s why multicellular organisms have evolved networks of chemical signalling that allow their cells to communicate with each other. A commonly observed feature in ageing is that these networks are gradually disrupted.
This disruption happens in several ways, such as increased inflammation levels, decreased effectiveness of the immune system against pathogens or potential cancer cells, and changes in the composition of cellular environments.
Chronic inflammation is perhaps the most prominent of these disruptive features, and it is so specific to ageing that it has earned the name “inflammaging”. According to The Hallmarks, the causes of inflammaging are multiple. Tissue damage causes inflammation, which in turns calls in repair mechanisms to fix the damage; unfixed (or unfixable) damage stays put, causing inflammation to continue. Similarly, infections cause inflammation; the immune system is supposed to notice it and swoop in to eliminate the threat, but as its effectiveness goes downhill in ageing, infections stick around for longer, and so does the resulting inflammation. Senescent cells, as we’ve seen, spew out pro-inflammatory chemicals, and as they grow in number over time, the inflammation they cause increases too. The list goes on, and so does the list of disorders commonly observed in ageing that are directly or indirectly linked to chronic inflammation: obesity, type 2 diabetes, bone fragility, muscle weakness, atherosclerosis, et cetera.
The Hallmarks cites a few studies that suggest possible ways to restore proper intercellular communication (or at least improve it), which include both nutritional and pharmacological interventions. As banal as it may sound, if the problem is chronic inflammation, the long-term administration of anti-inflammatories may help; altering the composition of the gut microbiome is a possible avenue too, as it is known that the microbiome influences the immune system, which as we’ve seen plays a role in how long inflammation lasts. Dietary restriction regimens may help do that, but that said, don’t go and pop an aspirin a day or start turning your eating habits on their head. Most, if not all, of these findings are still well within the realm of preliminary research, and shouldn’t be taken for granted until sufficiently large clinical trials prove them safe and useful.
Is that it? No.
That was a long read, and if you actually managed to keep your attention from drifting away, kudos to you. In that case, though, you might have noticed a pattern in the hallmarks. Some of them are presented as good for you to a point. Take cellular senescence: it prevents damaged cells from spreading, but when senescent cells become too many, they cause more harm than good. We saw a similar thing in the case of deregulated nutrient sensing, when weakening anabolic pathways too much does more damage than it prevents.
Other hallmarks too can be categorised in some way, and that’s actually what the authors of the paper did. They subdivided the hallmarks into primary, antagonistic, and integrative.
Primary hallmarks—genomic instability, telomere attrition, epigenetic alterations, and loss of proteostasis—are proposed as the causes of the damage that accumulates with age. Antagonistic hallmarks—mitochondrial dysfunction, cellular senescence, and deregulated nutrient sensing—are the good-to-a-point hallmarks, responses to damage that are good in the short term but bad in the long run. (After all, evolution cares about you only until reproductive age.)
The hallmarks of ageing are subdivided into primary, antagonistic, and integrative. (Source: The Hallmarks of Aging.)
Integrative hallmarks—stem cell exhaustion and altered intercellular communication—are the tip of the iceberg, the end result of damage and responses to damage, the last link in the chain connecting damage itself to the decline of health we see with age.
That’s what the authors of The Hallmarks propose, anyway, and as interesting as all of this may be, not all scientists agree that the paper nailed it. I’m not talking just about this subdivision in three categories; some are unconvinced that the hallmarks constitute a paradigm for ageing research. In other words, they aren’t sure that The Hallmarks is a valid theoretical model that accurately represents or explains the real-life phenomenon of ageing.
For example, in a recent critique, David Gems and João Pedro de Magalhães suggested that, while The Hallmarks has quietly passed for an explanatory framework for ageing, it’s more like a list of relevant topics within the field of ageing research. The critique points out how The Hallmarks of Aging attempted to do for the ageing field what The Hallmarks of Cancer did for oncology. This latter paper successfully built an explanatory framework for cancer that is supported by evidence, relying on the universally accepted notion that mutations are the primary cause of cancer. In a similar way, The Hallmarks of Aging assumes that the accumulation of damage is the primary cause of ageing that sets everything in motion, but this assumption is far from undisputed by other scientists. In addition, Gems and de Magalhães maintain that the list of hallmarks is somewhat arbitrary, and that it may well have included other possible hallmarks.
They also criticise the subdivision into primary, antagonistic, and integrative hallmarks, saying that it’s tentative and not supported by evidence. (As a side note, to a point, I got this impression as well: I can certainly see why cellular senescence is antagonistic, but I can’t see why mitochondrial dysfunction would be. I can see why the production of ROS that supposedly contributes mitochondrial dysfunction can be antagonistic, but the dysfunction itself? Then again, I’m no biologist and my opinion is therefore highly uneducated.)
Furthermore, Gems and de Magalhães said that the link between primary and antagonistic hallmarks isn’t clear enough, as isn’t how ageing itself would arise from the antagonistic and integrative hallmarks. (It certainly isn’t clear to me.) They concede that the paper is a great introduction to the field of ageing research, but they also express concern that taking it as a paradigm may carry the risk of undermining the development of a real paradigm that could allow us to fully understand and, hopefully, control ageing one day.
Give Caesar what is Caesar’s
Are the hallmarks an explanatory framework for ageing or not? If they aren’t, how close are they? I certainly don’t have an answer to that, but I think The Hallmarks has certainly contributed to legitimising ageing research—which has historically been very controversial at best—and has generated a lot of enthusiasm around it. That is part of the reason why the field has been growing so much faster in the past decade or so, but it would be unfair to say that the merit is all of this one paper.
I first approached this field about two years before The Hallmarks were published. What sparked my interest was SENS, an idea devised and championed by Aubrey de Grey in the early 2000s and described in his book Ending Aging. In its time, SENS—which stands for Strategies for Engineered Negligible Senescence—was extremely controversial, challenged, and even ridiculed, in spite of the fact that it’s conceptually not far at all from The Hallmarks.
Like The Hallmarks, SENS hinges on the assumption that ageing is ultimately a consequence of damage accumulation. Its proponents argue that, if we could fix that damage, we could slow down and possibly even reverse ageing. If we could do that, we might even achieve something close to negligible senescence, a phenomenon that occurs naturally in a few species and that basically boils down to their ageing being so slow to be virtually irrelevant, if at all present.
Instead of nine hallmarks, SENS lists only seven categories of cellular and molecular damage, for each of which it proposes a specific repair approach. SENS doesn’t really bother with trying to understand how damage happens, but only with how to fix it once it already has happened—the rationale being that we know nowhere near enough to prevent the damage from happening, but that we do know enough to try to fix it. (Again, not everyone agrees with this.)
Aubrey de Grey’s famous list of the “seven deadly things.” (Source: sens.org)
Even at glance there’s quite some overlap with The Hallmarks; I’m not going to go into details to avoid the irony of my readers dying of old age as they try to finish this article. The reason why SENS (and de Grey) were unjustly ridiculed are probably three: a) although he does have a PhD in biogerontology, de Grey didn’t follow a traditional academic path; b) de Grey is on the record for saying that we’ve got a 50-50 chance to be able to fix ageing in two decades or so, and that the first human to live to 1000 is probably already alive; c) the beard. I love it, but some people are just that thick and equate an unusually long beard with lack of credibility.
The 1000-year lifespan thing makes a lot of people choke on their coffee, but I don’t think it’s so far-fetched. One thing is being sceptical that we’re a mere two decades away from being able to fix ageing; many scientists aren’t convinced of that either, even some who think that we will eventually fully figure out and control ageing. Thinking that people just can’t live to a 1000 simply because it sounds outlandish is another thing, and it makes no sense at all. In the absence of ageing, there’s no reason to think you would spontaneously drop dead anyway when you’re about 80 just because you’re about 80. People who drop dead at 80 do so because, broadly speaking, their health has deteriorated too much for them to be alive. If you take ageing out of the equation, the length of your life depends only on how likely you are to die of other causes, like accidents, murder, infectious diseases, et cetera—all things we’re already very good at preventing, which is why life expectancy has skyrocketed in recent times. Ageing is kind of the big thing left to tackle.
I don’t know if we’re as close to fixing ageing as de Grey thinks, but I do believe that his work and his relentless advocacy efforts of the past twenty years have contributed a lot to popularising ageing research and making it the legitimate scientific field it is today. “Rejuvenation” was basically a forbidden word when he first started using it; now you routinely find it in research papers. So, if he’s right and if you’ll be a thousand years old one day, I think you’ll owe that to him in no small part. (And to many, many other scientists and advocates, for sure.)
Wrapping up
This post is part of the RJ files, a subsection of Too Many Things dedicated to ageing and life extension. At the time of writing, that section is very much a work-in-progress, but more posts will appear in the near future—particularly rebuttals to typical objections to life extension, though you can expect more posts about the science every now and again, if anything especially interesting attracts my attention.
If you’re eager to read more now, I do have an older article that summarises pretty much all I have to say about life extension. Be warned, though: it’s long and contains a fair amount of sarcasm. If that doesn’t scare you off, I wish you a pleasant reading.
In this second-to-last video on the hallmarks of ageing, we’ll see very briefly what stem cells are, and how their dwindling numbers over time contribute to making us grow older. Next week, I’ll publish the final hallmark video, altered intercellular communication, and hopefully I’ll also have a longer hallmark explainer article to go with it.
These videos are the reason why I haven’t been writing a proper post in ages, but rest assured I have more stuff in the pipeline, and not just about ageing and life extension. Some things are already in the works; others will be after a short break that I really need to take, otherwise I’ll blow all my own cells, stem or not.
Here’s the seventh episode in my hallmarks of ageing series: deregulated nutrient sensing. Cells are living things, and as such, they need to eat to live; they get their nutrients from the food we eat. (Well, yes, basically we eat to feed our cells.) Cells can tell how abundant or scarce the nutrients around are, but like all good things, this ability diminishes with ageing. Pathologies like type-2 diabetes are deeply intertwined with this hallmark, but I’ll leave the details for my hallmarks of ageing article, which if everything goes according to plan, I should publish around two weeks from now. That’s when the hallmarks of ageing series will be concluded.
Speaking of the end of this video series, the next video right after that will be about something else entirely. All I’m going to say is that it will be about a video game character, and it will likely be long. (Not for nothing I’ve been working on it since April.) Curious? Stay tuned, and for the time being, enjoy this short video.
My sixth video on the hallmarks of ageing focuses on cellular senescence, the hot topic of ageing research of the past decade. Cellular senescence is something that happens to cells when their DNA is so damaged that it’s better if they aren’t allowed to make copies of themselves anymore. Senescence is a kind of “undead” state for cells, and just like the undead, senescent cells have a thing for coming back to haunt the living.
There are only three more hallmarks of ageing left, at which point my video efforts will go quite a different direction (but I won’t spoil it for you just yet). If you landed on this blog (or my channel) because you’re interested in aging research and longevity, I guess this is a good moment to remind you that those aren’t the only topics that I like talking about. So, don’t be disappointed if one day you come here and see that I’m talking about videogames. (Which I’ve done more than once before.)
Still, if you don’t mind some variety, I invite you to take a look at my channel, and maybe subscribe.